Ⱦ�Ϸ�ˮ��������-������
����1 ����(Introduction) ����Ⱦ�Ϸ�ˮ����Ϊ�Ǽ��Ѵ�������Ⱦ��֮һ����֯����ֽ��ӡˢ��ʳƷ�ͻ�ױƷ������Ⱦ����Ⱦ����ŷ�.Ⱦ�Ϸ�ˮ���л��ﺬ���ߡ����Դ��ѽ���ȣ��ᵼ�����༲�����簩֢��ƶѪ�ͺ����ϰ���.Ŀǰ��Ⱦ�ϵĴ��������е绯ѧ����������������������Լ���������.�������������µ���Ⱦ��ܺĵͶ����ܹ�ע�����дŷ����������ŵ����ͻ�����ڼ̳����������ŵ��ͬʱ���ڷ��룬����ȥ����Ⱦ��. ������Ͱ���һ�ֶ������������ĵ��ʣ�Ҳ��һ�ּ���.�������𤸽������Ҫ�ɷ־��Ƕ�Ͱ�.����������Һ�����£���Ͱ���¶�ڿ����оͻ�ܿ���Ծ۲��������ɷۺ�ɫ�ľۺ϶�Ͱ����Ƚ��ջ�۳�Ϊ���з���ṹ�Ͷ���ӵ����壬�����������ȫ�Է��ģ���������Ծۺ��ڶ��ֻ��ʱ���.�ۺ϶�Ͱ�(PDA)�������õ���ˮ�ԡ����������Ժ��ȶ��ԣ����Ա�����������.�����Ժ���������������ʣ����ҿ��Էḻ�������ʣ�����������õ���Ӧ��.�����ö�Ͱ�����̼���ܣ������������������������ö�������Ͷ�Ͱ������ڴ������ײ����ϣ�Ӧ����������뷽��.Ȼ����PDA�����㲻�ȶ������״ӻ������Ѹ�����.����������ڸ��������������¸��¼��ȣ����Խ�PDA̼�����õ������ȶ���̼�������������������. �������о����þۺ϶�Ͱ�����������������Ȼ���ڸ�����̼���Ʊ�̼���ϰ�����������(Fe3O4@C).�Լ���ΪĿ����о��ò��ϵ���������. ����2 ʵ�鲿��(Experimental section)2.1 ʵ���Լ� ������ʵ�������Լ���Ϊ������.FeCl3·6H2O�ͼ��̹�������д�ï��ѧ�Լ���(�й������).���ǻ���������(Tris)���ڳɶ���ѧ�Լ���(�й����Ĵ�).�Ҷ������Ҵ�������̨���ͻ�ѧ�Լ�����˾(�й�����̨). ����2.2 �����Ʊ� ����Fe3O4���������������Ʊ�.ȡFeCl3·6H2O 2.7 g��NaAc 7.2 g�����Ҷ���2.0 g������80 mL�Ҷ����У����������壬Ȼ��ת�Ƶ����ķ���ϩ�ڳĵĸ�ѹ��Ӧ����200 ���·�Ӧ24 h��ȡ�������Ҵ���ϴ���Σ�����ˮϴ3�Σ��ô������룬���.ȡ200 mg Fe3O4��������240 mg Tris��240 mg ��Ͱ�����200 mL��ˮ�ڱ�ˮԡ�»�ϣ�����10 min�����½���24 h.��Ӧ��Ϻ��ô�ˮϴ��5�Σ��ŷ������.������ڹ�ʽ¯�����������600 ������4 h�������¹����п�����������Ϊ1 ��·min-1���Ƚ��յõ�Fe3O4@C.�ۺ϶�Ͱ�̿����(C)���Ʊ�������Һ�в�����Fe3O4�������⣬����������Fe3O4@C���Ʊ�����. ����2.3 ���ϱ��� ����������羵(TEM��H-7500��Hitachi��Japan)��ɨ��羵(SEM��SUPPATM55��Zeiss��Germany)�۲��Ƶõ��������ϵ���ò���������������ϵĴ����ܲ�����ǿ��(VSM��LDJ9600)���.���ø���Ҷ�任���������(FT-IR NEXUS 670 Madison��WI��USA)�����������.���ϵľ���ṹ����X�������������(Rigaku D/max2500 VPC���ձ���ѧ)�ⶨ.Ϊ����̿���ϵĽṹ�����ü���۽�����������(Lab RAM HR800������Horiba Jobin Yvon��˾)����Ʒ���з��������ü��Ⲩ��Ϊ530 nm.���ϵıȱ��������϶���Բ��ñȱ��������϶�ȷ�����(ASAP2020��������˹�˾)�ⶨ. ����2.4 �������� ��������������100 mL����ϩƿ��װ25 mL��Һ.��������Ũ��Ϊ0.20 g·L-1������Ũ�ȴ�20~140 mg·L-1������������ʵ�飬ѡ��ˮ��ȡ��У�ĺ�ˮ��ʵ���ҵ�����ˮ��Ϊ���ʿ��첻ͬˮ������������.pH���ڴ�3.0~10.0���о���ȶԼ���������Ӱ��.�������Һ��30 �����ˮԡ���ڲ�ͬ�����ʱ�����ȡ�����ⶨ���̵���������ѧ.����ѧʵ��ѡ�������¶�Ϊ20��30��40 ��.��������ʱ�����Һ���ڴ����Ͻ��з���5 min��Ȼ��ȡ�ϲ���Һ������ֹ��ȼ�(UV-2550��Shimadzu)���ⶨ���̵�Ũ��.������ʵ��ƽ�н���3�Σ����б�������Ϊ3��ƽ��ֵ. ����3 ���������(Results and discussion)3.1 ���ϱ������ ����ͼ 1aΪFe3O4ɨ��羵ͼ�����Կ���Fe3O4���η�ɢ���ȣ�ֱ��ԼΪ160~300 nm��������濴�����ֲڣ��ѷ��.ͼ 1bΪFe3O4@C���ϣ�����⻬�ѷ��ϸ�¾��ȣ�ֱ��Ҳ����ԼΪ330~390 nm.��羵ͼ 1c��ʾ��Fe3O4Ϊ����ɢ���������������������ȣ�ԼΪ200 nm.ͼ 1d Fe3O4@C����ͼ�п��Կ������ڲ���ɫ�Ŀ���ΪFe3O4������������ɫ�IJ����ǰ����ۺ϶�Ͱ����պ��̼����Լ50 nm���������ȣ���������.ɨ��羵����羵ͼ��֤����Fe3O4����ȫ��������̼�Dz���.�ȱ��������϶�ȷ����Dz��Fe3O4@C���ϵ�BET�ȱ����Ϊ98.83 m2·g-1�������Ϊ0.064 cm3·g-1��ƽ����Ϊ25.9 nm��Fe3O4@C�����ϵĿ�϶�Խ��Ϊ������Щ��ṹʹ�ò��Ͼ��бȽϸߵıȱ�����Ϳ������������������������ܣ���ɨ��羵ͼ��Ҳ���������Ĺ۲쵽���������̿���ϵĿ�϶. ���� 
����ͼ 1 Fe3O4��Fe3O4@C��ɨ��羵(a��b)����羵(c��d)ͼ ����ͼ 2������Fe3O4��Fe3O4@C�Ĵ��ͻ���.Fe3O4û�д�����������Ժ�ʣ�Ż���Ϊ�㣬���ֳ�˳���Ե����ԣ���Fe3O4@C���������ԵĴ�������ʣ��Ϊ7.86��������Ϊ251����ʾ�����������ԣ�������������ھۺ϶�Ͱ�̿���Ĺ����в���Fe3O4�ڸ������������ΪFe2O3.Fe3O4��Fe3O4@C�ıȽϴʹ�ǿ�ȷֱ�Ϊ78.7 emu·g-1��47.2 emu·g-1.Fe3O4@C���Խ�������Ϊ���������ԵĿDz�.������������Ⱦ������ô��������״���Һ�з������. ���� 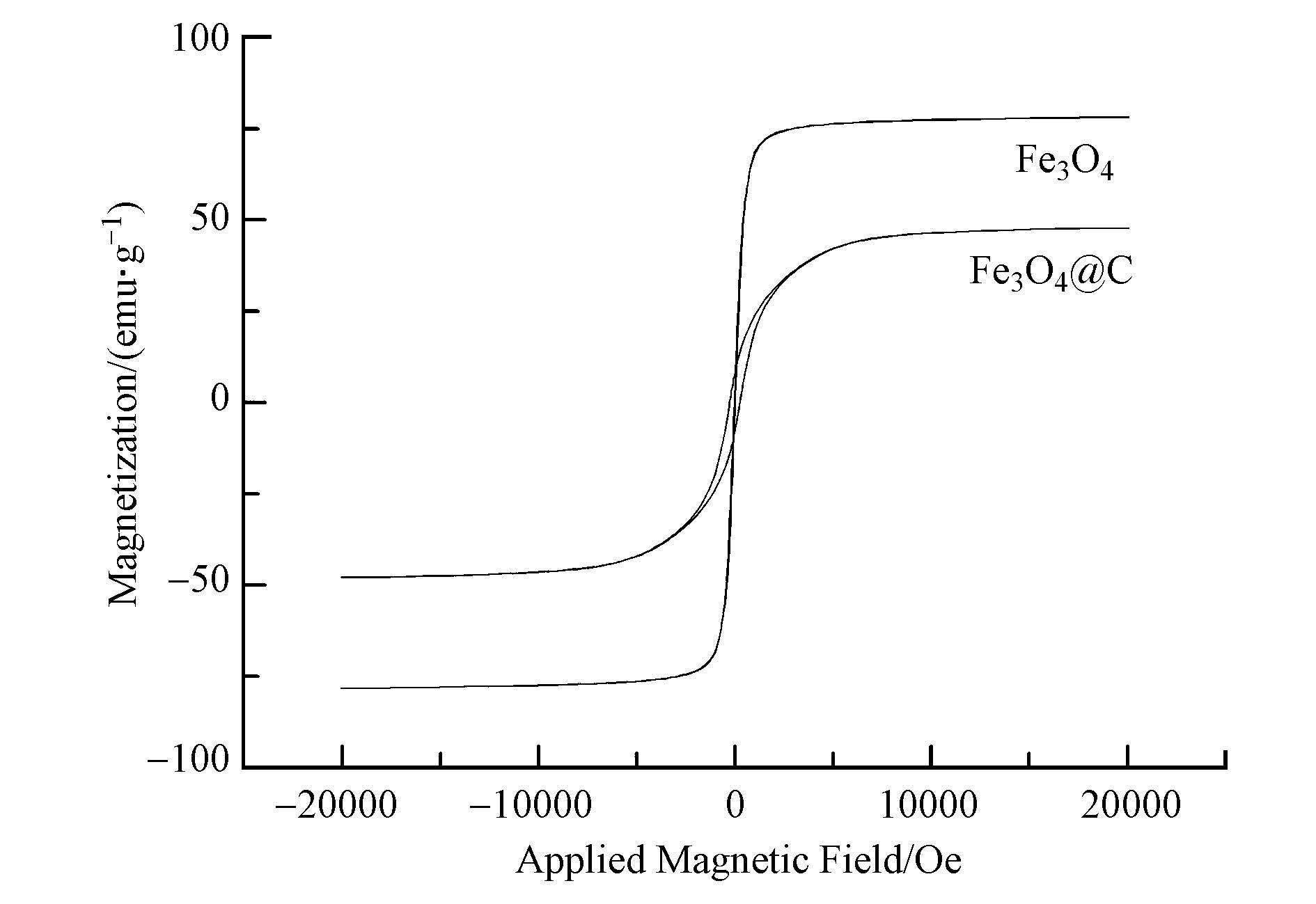
����ͼ 2���ϵĴ��ͻ��� ����ͼ 3������Fe3O4���۶�Ͱ�������Fe3O4@PDA��Fe3O4@C��X��������ͼ.Fe3O4��Fe3O4@PDA������λ����ȫһ������2θΪ30.12°��35.38°��43.05°��53.39°��56.91°��62.54°�������˽�ǿ������壬��ֱ��Ӧ������Fe3O4��(220)��(311)��(400)��(422)��(511)��(440)����(�ŠD�ȣ�2011;Σ���ȣ�2012;�Ʊ����ȣ�2012)�������ڸ�����̿���õ���Fe3O4@C��������λ����������⣬��2θΪ33.17°��49.33°���н�С������壬��Ӧ��б����Fe2O3��(104)��(024)���棬��˵����̿���Ĺ����в���Fe3O4���ΪFe2O3������Բ����Ľ��һ��. ���� 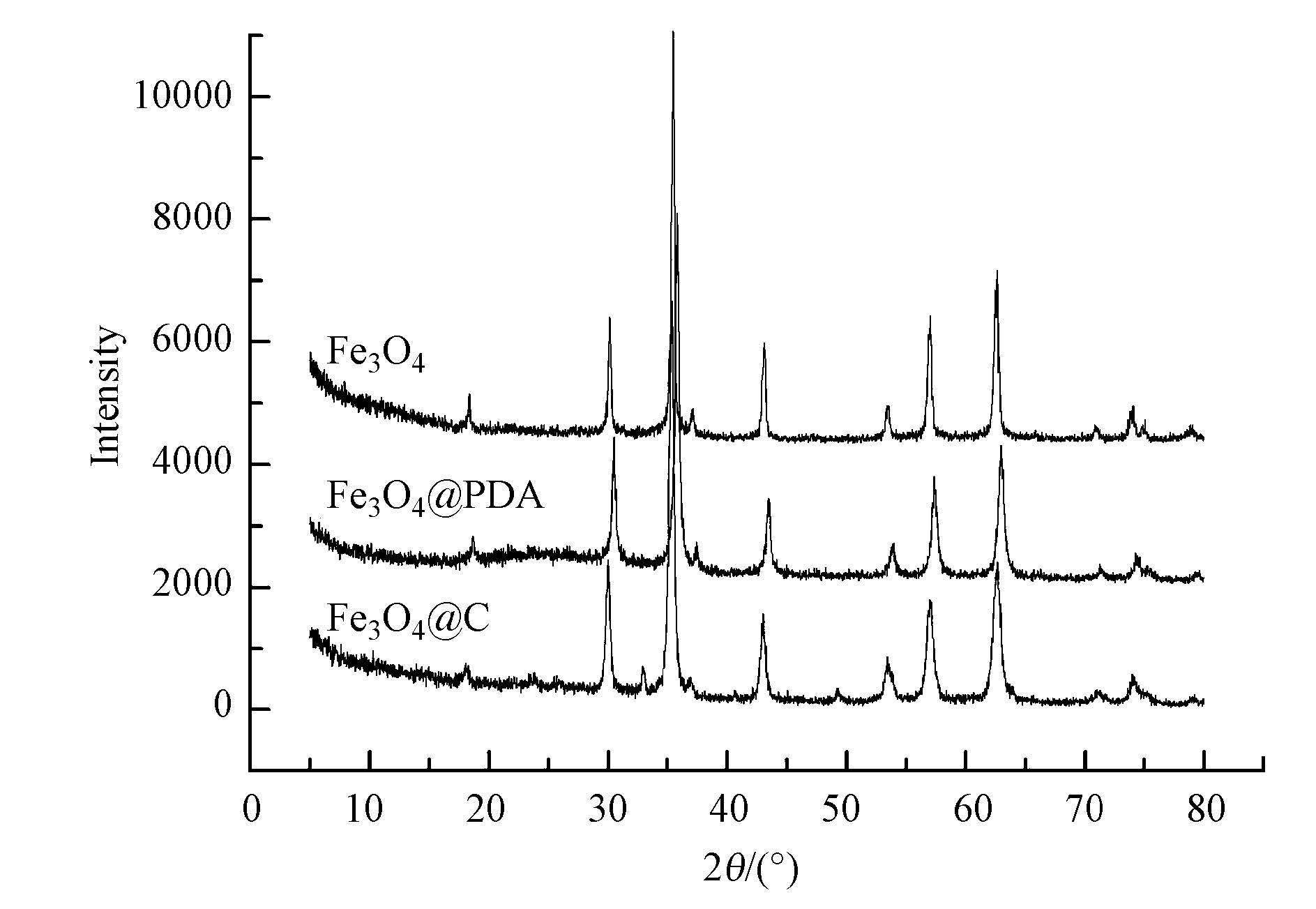
����ͼ 3���ϵ�X������������ ���� 
����ͼ 4 Fe3O4��Fe3O4@C��C�ĺ������ͼ ����ͼ 4��Fe3O4��Fe3O4@C��C�ĺ������ͼ��ͼ�о���3440.98 cm-1��1638.86 cm-1���������շ壬�ֱ�Ϊ���ϱ�������ˮ��O—H���������������(��ѩ�ȣ�2013)��̿����IJ�����3440.98 cm-1���ķ�ǿ�����Խ��ͣ��������ڲ���̿������ˮ����ǿ����������ˮ������ɵ�.Fe3O4��Fe3O4@C����ͼ��584.89 cm-1���ֵ����շ���Fe—O�������壬���Ұ�����C���Ϻ�Fe—O�ķ����Լ�С.��Fe3O4����ͼ��2844.59��2926.20 cm-1���ֵ�С��Ϊ—CH2��—CH3��C—H��������(Wang et al��2008)����ЩC—H������������������ܼ��ȷ��Ʊ�Fe3O4�����������л��ܼ������.�ۺ϶�Ͱ�̿����IJ��Ϻ�����ͼ��ȥû��Fe—O���������⣬������Fe3O4@C����ͼ���ƣ��������̿���ϱ�������Fe3O4��. ����ͼ 5ΪFe3O4@C����������ͼ��ͼ���г����ڲ���Ϊ1350 cm-1�����ķ��ΪD�壬D�巴ӳ̿ԭ�Ӿ����ȱ�ݳ̶ȣ���ʯī����ȱ�ݡ���Ե�������к͵ͶԳ�̼�ṹ������ģ�����̿�������Ҳ�ṹ��������̼��Ӧ�������壬��ǿ�ȱ���������sp2̼��ɵ�������ƽ���Լ���ʯī���߽������������ȣ�λ�ڲ���Ϊ1580 cm-1�����ķ�ΪG�壬����ʯī����������������Ͳ��ϵ�ʯī���̶��йأ�����������ʯī�ķ�(�������ȣ�2013;�ܻ����ȣ�2014;��ΰ�ȣ�2014).D���G��ij��ֱ���̿���ϳɹ��İ�����Fe3O4�ϣ�G��ǿ��ǿ��D��˵���ڸ���̿���Ĺ����в���̿���ϱ�ʯī��. ���� 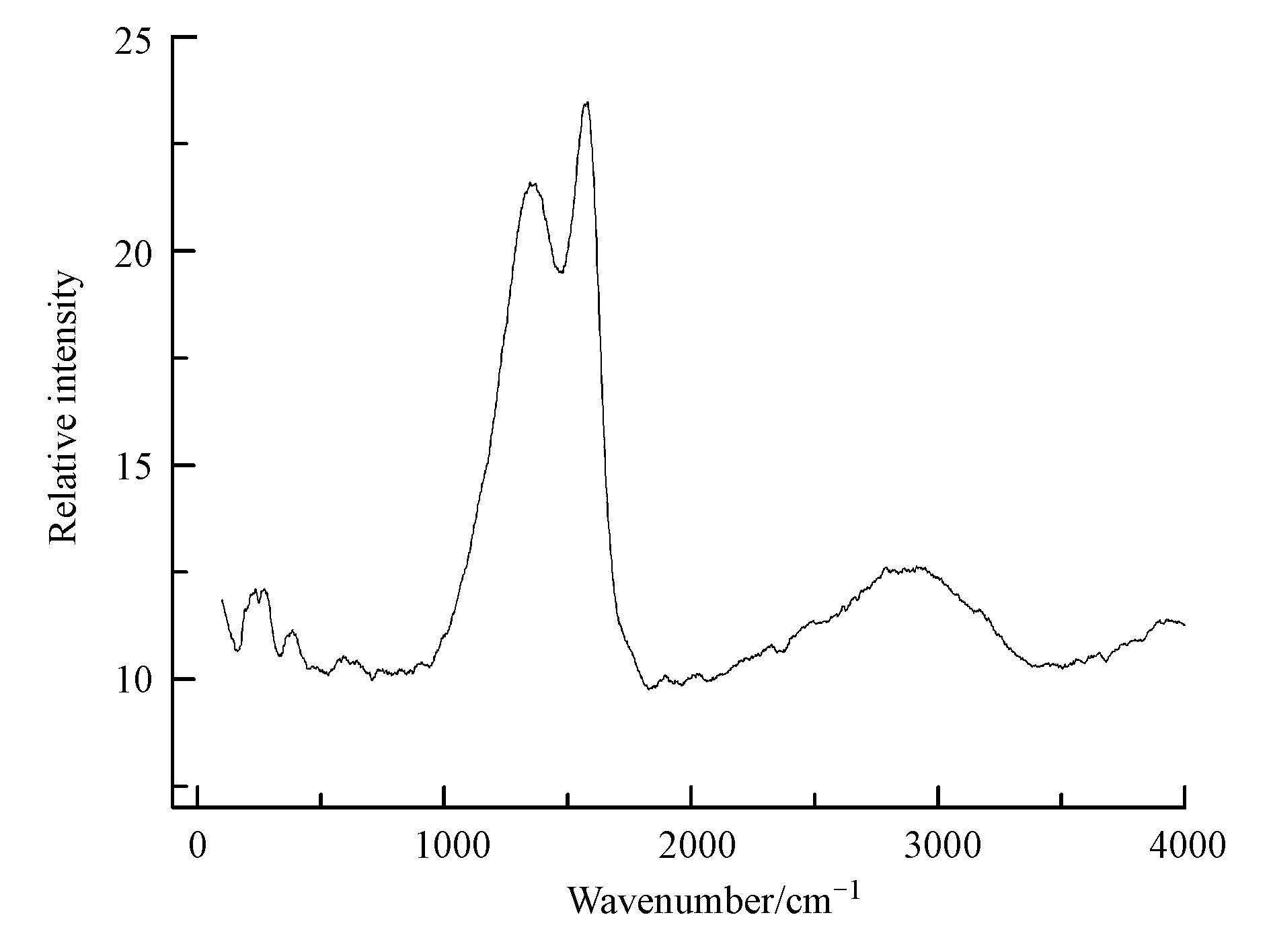
����ͼ 5 Fe3O4@C���������� ����3.2 ��ҺpHֵ��������Ӱ�� ������Һ��pH���������Ľ��λ���Լ�����Ӱ��ܴ�����о���ͬpH���������Լ��̵�����.��ͼ 6��ʾ���ڵ�pH�¼��̵���������С������pH���ӣ�������Ҳ���ӣ���pHΪ8��ʱ���������������ڲ���.�ڵ�pH�£��������������ӻ�����������ɣ�����������Һ���������ӵ���ʽ���ڣ�����֮��ľ���������²�����Ч������.pH�����Fe3O4@C���������Ӵ����磬�ھ��������£�������������Ӵ�����һ������.����Fe3O4@C�Լ��̵ĺ�������ʵ���趨�����Ժ������������½���. ���� 
����ͼ 6��ҺpH��Fe3O4@C�������̵�Ӱ�� ����3.3 ���������� �������������о���30 ���½��У����̳�ʼŨ������Ϊ20��40��60��80��100��120��140 mg·L-1��Ϊ�����������Բ�ͬˮ���м��̵�����Ч����ѡ��ˮ����ˮ������ˮ��Ϊ���ʣ�����������ͼ 7a.���Ÿ�ˮ���м���Ũ�ȵ����������������ڿ�ʼʱ������������������ƽ����˵�������ﵽ�˱���.�����ʸ��Ѷ�������ϣ����ģ�Ͷ��������ݽ�����ϣ�����ģ�����Է�����(1)��(2)��ʾ. ���� 
����(1) ���� 
����(2) ����ʽ�У�qe��Ce �ֱ�Ϊƽ����������(mg·g-1)��ƽ��Ũ��(mg·L-1);θΪ�Ƚϴ�����������bΪƽ����������.�Ƚϴ���������θ����ͨ��Ce /qe��Ceб�ʵ����� ͼ����.KF(mL1/n μg1-1/n)��n��Freundlich����.n��KF��ֵ��ͨ��logqe��logCe��������ϵĽؾ����.ͼ 7b���������ݵ�Langmuir�������. ���� 
����ͼ 7 Fe3O4@C�Լ��̵�����������(a)��Langmuir�������(b) ������ 1������ģ�͵���ϲ���.��ͬһˮ����Langmuirģ����ϵ����ϵ��(R2)Ҫ����Freundlichģ�ͣ��������ݺ�Langmuirģ������ϣ�˵��Fe3O4@C�Լ��̵�������һ�ֵ����Ӳ�����.ʹ��Langmuir�������������Fe3O4@C�Դ�ˮ����ˮ������ˮ�м��̵ıȽϴ����������ֱ�Ϊ490.1��442.5��389.1 mg·g-1.�ʹ�ˮ��ȣ���ˮ������ˮ�����������������ͣ���������������к��еĸ���������ɵģ�����Ȼ�������൱�ߵ���������.�� 2�г��������б������������Լ��̵��������������Կ���������ԭʯīϩ���������ϸ��⣬���о��Ʊ���Fe3O4@C�����������������������������Ҹ��������������ô�������ʵ�ֹ�Һ������ŵ㣬˵�����������ڴ���Ⱦ����Ⱦ�﷽���нϴ��DZ��. ���� 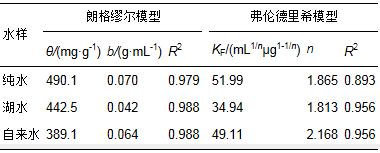
������ 1 ������Fe3O4@C��������Langmuir��Freundlichģ����ϲ��� ���� 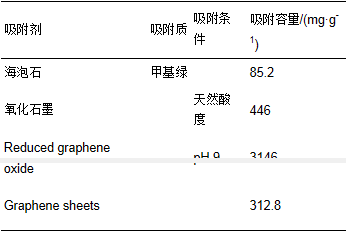
������ 2 ���ױ����������������Լ��̵��������� ����3.4 ��������ѧ �������̵���������ѧ��ͼ 8a�����̵���������ʱ������ӿ�����������һ��ʱ�������ƽ��������ʱ��ﵽ200 min������������������ʱ���ӳ������ӣ�˵���Ѿ��ﵽ����ƽ��.�����������ѧģ������Fe3O4@C�Լ��̵����������Է�����ʽ(3)��(4)��ʾ. ���� 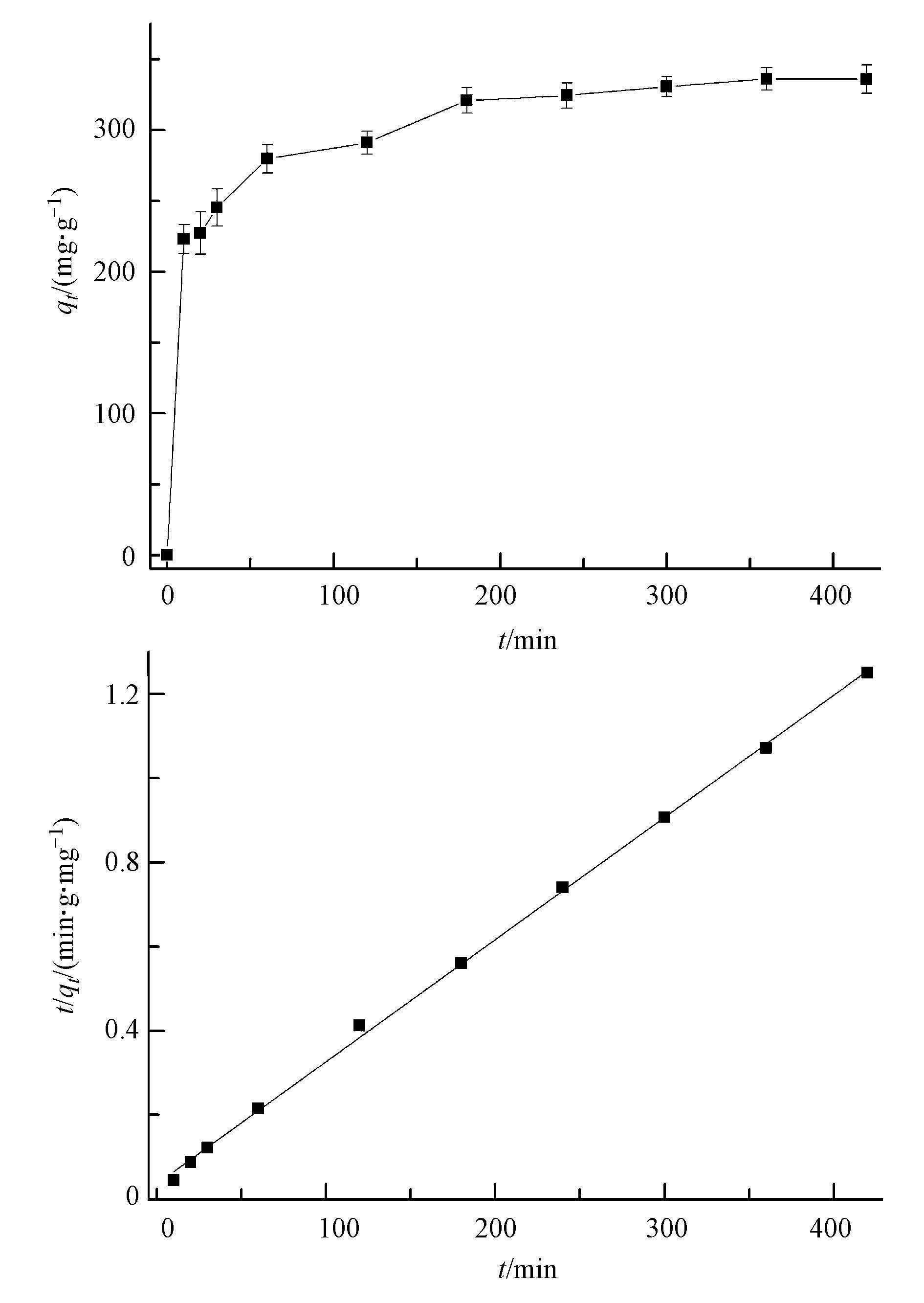
����ͼ 8������Fe3O4@C��(a)��������ʱ��ı仯��(b)���������ѧ������� ���� 
����(3) ����ʽ�У�k���������ʳ���(g·mg-1·min-1)��qt������ʱ�̼��̵�������(mg·g-1)��qe��ƽ��������.��ʼ��������h(mg·g-1·min-1)���������·���ʽ������ ����
����(4) �����������������ѧ�����ͼ 8b��ʾ���������ã��ɾ�ϵ��R2��0.999����.k��h����ͨ��ʵ�����ݣ�t/qt��t��ͼ��ֱ��б�ʺͽؾ�ȷ��.��������������ϵĿɾ�ϵ��R2����0.998���������������ѧģ��.��ͼ��б�ʺͽؾ�������������ʳ���k����ʼ��������h���ڱ� 3. ���� 
������ 3 Fe3O4@C�Լ��̵��������������ѧ���� ����3.5 ��������ѧ�о� �������̵ij�ʼŨ���趨Ϊ100 mg·L-1����������Ũ��Ϊ0.2 g·L-1���ı������¶�Ϊ20��30��40 �棬����ʱ���趨Ϊ6 hʹ��ﵽ���ƽ�⣬�ⶨ����Һ��ʣ����̵�Ũ��Ce��������������qe����ͬ�¶��µ������������ 4��ʾ.����ѧ������������(Biswas et al��2007)������ѧ���̽��м��㣺 ����
����(5) ����
����(6) �����ϲ���ʽ(5)��(6)���ù�ʽ(7)�� ���� 
����(7) ����ʽ(7)�У�KcΪ�¶�T�µ�ƽ�ⳣ��������ͨ��Kc=qe/Ce���㣬qeΪƽ������������CeΪ��Һ�������ʵ�ƽ��Ũ�ȣ�ΔGθ��ΔHθ��ΔSθ�ֱ�Ϊ��״̬��������Ӧ�ļ���˹�����ܱ䡢�ʱ���ر�.��ʽ(7)Ҳ���Ա�ʾΪ�� ���� 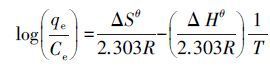
����(8) �����趨ΔHθ��ΔSθ���о����¶ȷ�Χ��Ϊ���������ڹ�ʽ(8)�У���(qe/Ce)��(1/T)����������ϣ���б�ʺͽؾ�������ΔHθ��ΔSθ����ͬ�¶��µ�ΔGθ���ɹ�ʽ(5)��ã�������ѧ�������� 4. ���� 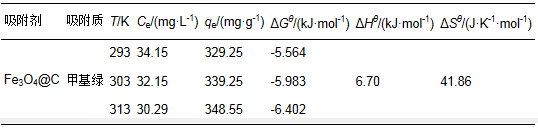
������ 4 ��ͬ�¶��µ�ƽ��Ũ�ȡ���������������ѧ������ �����ɱ� 4������ѧ����ֵ���Կ�����ΔGθС���㣬˵��������һ���Է��Ĺ��̣��������¶�����ΔGθ��С��˵���¶�Խ�ߣ��Է���Խǿ;ΔHθ�Ǵ����������˵�������������ȹ���;ΔSθ������˵����������һ�������ӵĹ���.����μ���ˮ���̳����ϻ�http://www.dowater.com������ؼ����ĵ��� ����4 ����(Conclusions) ��������ˮ�Ⱥϳɷ��Ʊ�Fe3O4�������ӣ�����һ��ۺ϶�Ͱ���Ȼ��̼���Ʊ�Fe3O4@C���ϲ��ϣ�������Ϊ����������ȥ������Ⱦ��.����ҺpHΪ���Ժ������������²��϶Լ��̾��нϺõ�����Ч��.��������ѧ���ݺ������������ݷֱ�������������ѧģ�ͺ��ʸ��Ѷ���������ģ�ͣ����нϸߵ���������.Fe3O4@C���ϲ��Ͼ���̼�����ȶ��Ļ�ѧ���ʺ��������������ܣ������ôų�ʵ�ֿ��ٷ��룬�ڻ�����ѧ��������ѧ�Լ����ﻯѧ���������һ����Ӧ��DZ��. ���������Զ����ֱ��!�Żݵļ۸�!�ܵ��ķ���! ����ˮ������Ʒ��ѯ���ߣ�010-8022-5898 186-1009-4262 (���α༭�����ܰ) |